7. 実践編: ディープラーニングを使った配列解析¶
![]()
近年,次世代シーケンサ(NGS; Next Generation Sequencer)の発展により,遺伝子の塩基配列が高速,大量,安価に読み取られるようになってきました.
ここではディープラーニングを用いて,DNA配列からエピジェネティックな影響や転写制御を予測する問題に取り組みます.ディープラーニングは複雑なモデルを表現でき,遠距離の影響も考慮することができ,より高い精度で予測することが期待できます.
7.1. 環境構築¶
ここで用いるライブラリは
- Chainer
- Cupy
- matplotlib
です.Google Colab上では,これらはあらかじめインストールされています.
以下のセルを実行して,各ライブラリのバージョンを確認してください.
[1]:
import chainer
import cupy
import matplotlib
chainer.print_runtime_info()
print('matplotlib:', matplotlib.__version__)
Platform: Linux-4.14.137+-x86_64-with-Ubuntu-18.04-bionic
Chainer: 6.5.0
ChainerX: Not Available
NumPy: 1.17.3
CuPy:
CuPy Version : 6.5.0
CUDA Root : /usr/local/cuda
CUDA Build Version : 10000
CUDA Driver Version : 10010
CUDA Runtime Version : 10000
cuDNN Build Version : 7603
cuDNN Version : 7603
NCCL Build Version : 2402
NCCL Runtime Version : 2402
iDeep: 2.0.0.post3
matplotlib: 3.1.1
期待される実行結果例
Platform: Linux-4.14.137+-x86_64-with-Ubuntu-18.04-bionic
Chainer: 6.5.0
ChainerX: Not Available
NumPy: 1.17.3
CuPy:
CuPy Version : 6.5.0
CUDA Root : /usr/local/cuda
CUDA Build Version : 10000
CUDA Driver Version : 10010
CUDA Runtime Version : 10000
cuDNN Build Version : 7603
cuDNN Version : 7603
NCCL Build Version : 2402
NCCL Runtime Version : 2402
iDeep: 2.0.0.post3
matplotlib: 3.1.1
7.2. 配列解析について¶
次世代シーケンサの発展・普及とともに,大量の遺伝子配列が読み取られるようになりました.そうした中で,塩基配列で表現された遺伝子型と病気や形態などの表現型との関係を推定するようなGWAS(Genome Wide Association Study; ゲノムワイド関連解析)が広がってきました.しかし,遺伝子の変異だけでは全ての表現型の変化を説明できないことがわかってきました.特に,非翻訳領域が遺伝子発現に影響を与え,表現型の変化を生じさせていることが様々な実験結果からわかってきています.遺伝子発現時に周辺領域がどのように影響を与えているのかを調べるために様々な手法が提案されています.
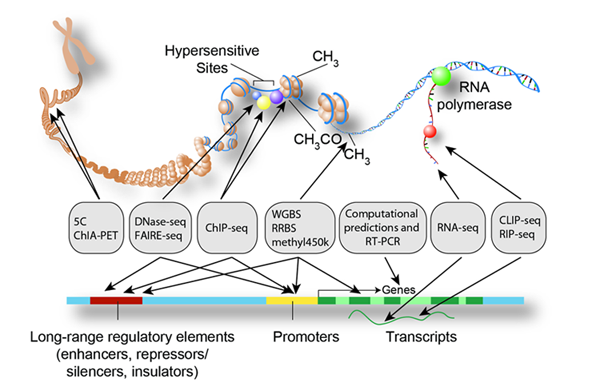
例えば,ChIP-seq(クロマチン免疫沈降シーケンス)は,ChIP(クロマチン免疫沈降)と高速DNAシーケンスを組み合わせることで,ヒストン修飾状況や転写調節因子の結合部位を網羅的(ゲノムワイド)に同定する手法です.これにより,転写調節機能を司るヒストン修飾やDNA結合タンパクの結合部位をゲノム全体で同定することができ,遺伝子変異だけでは説明しきれない細胞の表現型に関与する膨大な情報の取得が可能になります.
そこで本節では,ChIP-seqにより得られた転写調節因子の結合部位に当たるDNA塩基配列のパターンを深層学習により学習することで,任意のDNA塩基配列に対して特定の転写調節因子との結合可能性の予測を行います.このアプローチはゲノム全体のヒストン修飾部位の予測やオープンクロマチン領域の予測など幅広い生命現象を統一的に取り扱うことを可能とします。
この課題を機械学習で取り扱う際の技術的な難しさの一つが,DNA塩基配列の長距離相互作用と呼ばれる現象です.これは,核内のDNAは複雑に折り畳まれた様式で存在しており,塩基配列上の並びとしては遠く離れた2つの領域が空間的には近い距離に位置し,転写調節因子の結合に影響を及ぼすことがあるということです.例えば,今回対象とする問題では10万bp (ベース・ペア:DNAを構成する塩基を数える単位) 超の長さのDNA塩基配列を入力として受け取り,DNA塩基配列中のある領域が転写調節因子の結合部位になり得るかを予測します。このような長距離相互作用を考慮しても効率的に学習可能なモデルを構築してきます‥
今回は,数百種類の人の細胞型から得られた数千のChIP-seq,DNase-seq(オープンクロマチン領域の網羅的解析の一手法)のデータセットから得られたDNA塩基配列を入力として,CAGE(Cap Analysis of Gene Expression)の結果計測されたmRNAの発現量を推定する問題を考えます[1].
7.3. データセット¶
ここでは,Basenji[1]で使われた実験データセットの一部を利用します.これらはCAGEなどの配列解析処理を行って得られたデータセットです.
下のセルを実行してデータをダウンロードしてください.
この配列はそれぞれが長さ131072bpからなり,128bp毎に対しそのカバレッジ値が記録されています.このカバレッジ値の配列の長さは131072/128=1024です.
この問題の目標は長さ131072bpの配列を入力として受け取った時に,この128bp毎のカバレッジ値を推定することが目標です.
今回は10種類の異なる実験のカバレッジ値を同時に予測する問題を扱います.
[2]:
!wget https://github.com/japan-medical-ai/medical-ai-course-materials/releases/download/v0.1/seq.h5
--2018-12-16 04:41:34-- https://github.com/japan-medical-ai/medical-ai-course-materials/releases/download/v0.1/seq.h5
Resolving github.com (github.com)... 192.30.253.113, 192.30.253.112
Connecting to github.com (github.com)|192.30.253.113|:443... connected.
HTTP request sent, awaiting response... 302 Found
Location: https://github-production-release-asset-2e65be.s3.amazonaws.com/153412006/c79a0800-f713-11e8-8d6c-255563d45b1b?X-Amz-Algorithm=AWS4-HMAC-SHA256&X-Amz-Credential=AKIAIWNJYAX4CSVEH53A%2F20181216%2Fus-east-1%2Fs3%2Faws4_request&X-Amz-Date=20181216T044134Z&X-Amz-Expires=300&X-Amz-Signature=df390bc2ed4392cbdd65444198dcec236c19532e06158d89cda5c2fe4e17f5db&X-Amz-SignedHeaders=host&actor_id=0&response-content-disposition=attachment%3B%20filename%3Dseq.h5&response-content-type=application%2Foctet-stream [following]
--2018-12-16 04:41:34-- https://github-production-release-asset-2e65be.s3.amazonaws.com/153412006/c79a0800-f713-11e8-8d6c-255563d45b1b?X-Amz-Algorithm=AWS4-HMAC-SHA256&X-Amz-Credential=AKIAIWNJYAX4CSVEH53A%2F20181216%2Fus-east-1%2Fs3%2Faws4_request&X-Amz-Date=20181216T044134Z&X-Amz-Expires=300&X-Amz-Signature=df390bc2ed4392cbdd65444198dcec236c19532e06158d89cda5c2fe4e17f5db&X-Amz-SignedHeaders=host&actor_id=0&response-content-disposition=attachment%3B%20filename%3Dseq.h5&response-content-type=application%2Foctet-stream
Resolving github-production-release-asset-2e65be.s3.amazonaws.com (github-production-release-asset-2e65be.s3.amazonaws.com)... 52.216.137.212
Connecting to github-production-release-asset-2e65be.s3.amazonaws.com (github-production-release-asset-2e65be.s3.amazonaws.com)|52.216.137.212|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 594118876 (567M) [application/octet-stream]
Saving to: ‘seq.h5’
seq.h5 100%[===================>] 566.60M 71.1MB/s in 8.5s
2018-12-16 04:41:43 (66.7 MB/s) - ‘seq.h5’ saved [594118876/594118876]
[3]:
!ls -lh
total 567M
drwxr-xr-x 1 root root 4.0K Dec 10 17:34 sample_data
-rw-r--r-- 1 root root 567M Dec 3 06:54 seq.h5
seq.h5というファイルが正しくダウンロードされているかを確認してください.サイズは567MBです.
seq.h5はHDF5形式でデータを格納したファイルです.HDF5ファイルは,ファイルシステムと同様に,階層的にデータを格納することができ,行列やテンソルデータをそれぞれの位置で名前付きで格納することができます.
HDF5形式のファイルを操作するためにh5pyというライブラリがあります.h5pyのFile()関数でファイルを開き,keys()関数でその中に含まれているキーを列挙します.また取得したキーを'[]'内で指定することでそのキーに紐付けられて格納されている各データを参照することができます.
テンソルデータはnumpyと同様にshapeという属性でそのサイズを取得することができます.
以下のセルを実行して格納されているデータを確認してください.
各データの名前にtrain(学習),validate(検証),test(テスト)の接頭辞がつけられ,inが入力の塩基配列,outが出力のカバレッジ値に対応します.
例えば,'train_in'は学習用の入力データであり(5000, 131072, 4)というサイズを持ちます.これは長さが130172からなる配列が5000個あり,それぞれA, T, C, Gの対応する次元の値が1, それ以外は0であるような配列です.
また,'train_out'は学習用の出力データであり,('5000, 1024, 10')というサイズを持ちます.これは長さが1024からなる配列が5000個あり,それぞれが10種類の異なるChIP-seqの結果のカバレッジ値が格納されています.
[4]:
import h5py
import numpy as np
with h5py.File('seq.h5', 'r') as hf:
for key in hf.keys():
print(key, hf[key].shape, hf[key].dtype)
target_labels (10,) |S29
test_in (500, 131072, 4) bool
test_out (500, 1024, 10) float16
train_in (5000, 131072, 4) bool
train_out (5000, 1024, 10) float16
valid_in (500, 131072, 4) bool
valid_out (500, 1024, 10) float16
(u'target_labels', (10,), dtype('S29'))
(u'test_in', (500, 131072, 4), dtype('bool'))
(u'test_out', (500, 1024, 10), dtype('<f2'))
(u'train_in', (5000, 131072, 4), dtype('bool'))
(u'train_out', (5000, 1024, 10), dtype('<f2'))
(u'valid_in', (500, 131072, 4), dtype('bool'))
(u'valid_out', (500, 1024, 10), dtype('<f2'))
h5py形式のファイルをnumpyデータとして扱うには,コピーする必要があります.以下のコードは'train_in'というキーに対応するテンソルデータをnumpyデータとして読み出し,そのデータの一部を表示します.
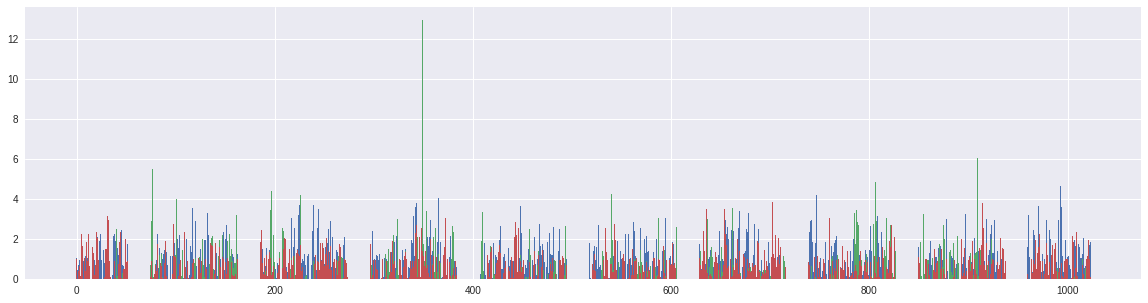
試しに最初のデータを取り出して,それの出力の値を表示してみます.
下のセルを実行してみてください.最初のデータの出力の三つの値を線グラフで出力します.(ここまでのセルを実行していてください).
[5]:
%matplotlib inline
import matplotlib.pyplot as plt
with h5py.File('seq.h5') as hf:
y = hf['train_out'][:100]
fig_size = plt.rcParams["figure.figsize"]
fig_size[0] = 20
fig_size[1] = 5
for i in range(3):
plt.bar(range(y.shape[1]), y[0,:,i])
7.4. Dilated Convolutionを用いた解析¶
7.4.1. 配列解析の戦略¶
今回は配列データが入力であるような問題である.
配列データを扱うためには大きく3つの戦略があります.
一つ目は,配列中の順序情報は捨てて,配列をその特徴の集合とみなすことです.これはBag of Words(BoW)表現とよびます.このBoW表現は特徴に十分情報が含まれていれば強力な手法ですがDNA配列のような4種類の文字からなる配列やその部分配列だけではその特徴を捉えることは困難です.
二つ目は配列中の要素を左から右に順に読み込んでいき計算していく手法です.これは4章でも少し触れたRNNを用いて解析します.RNNは時刻毎に入力を一つずつ読み取り内部状態を更新していきます.RNNの問題点はその計算が逐次的であり計算量が配列長に比例するという点です.現在の計算機は計算を並列化することで高速化を達成していますがRNNは計算を並列化することが困難です.もう一つの問題は遠距離間の関係を捉えることが難しいという点です.RNNはその計算方式から,計算の途中結果を全て固定長の内部状態ベクトルに格納する必要があります.遠距離間の関係を捉えようとすると,多くの情報を覚えておかなければなりませんが状態ベクトルサイズは有限なので,遠距離間の関係を捉えることが困難となっていきます.
三つ目は配列データを1次元の画像とみなし,画像処理の時と同様にCNNを用いて解析する手法です.CNNはRNNの場合と違って各位置の処理を独立に実行できるため並列に処理することができます.
今回はこの3つ目の戦略,CNNを用いて解析する手法を採用します.また,Dilated Convolutionを使うことで各位置の処理は遠距離にある情報を直接読み取ることができます.次の章でDilated Convolutionについて詳しくみていきます.
7.4.2. Dilated Convolution¶
従来の畳み込み層を使って配列解析をする場合を考えてみます. 以下の図のようにある位置の入力の情報は各層で隣接する位置からしか読み込まれません.どのくらい離れた位置から情報を取得するかはカーネルサイズによって決定され,カーネルサイズがKの時,Dだけ離れた距離にある情報を取得するためにはD/K層必要となります.今回の問題の場合Dは数百から数万,Kは3や5といった値ですので必要な層数も百から万といった数になってしまい現実的ではありません.
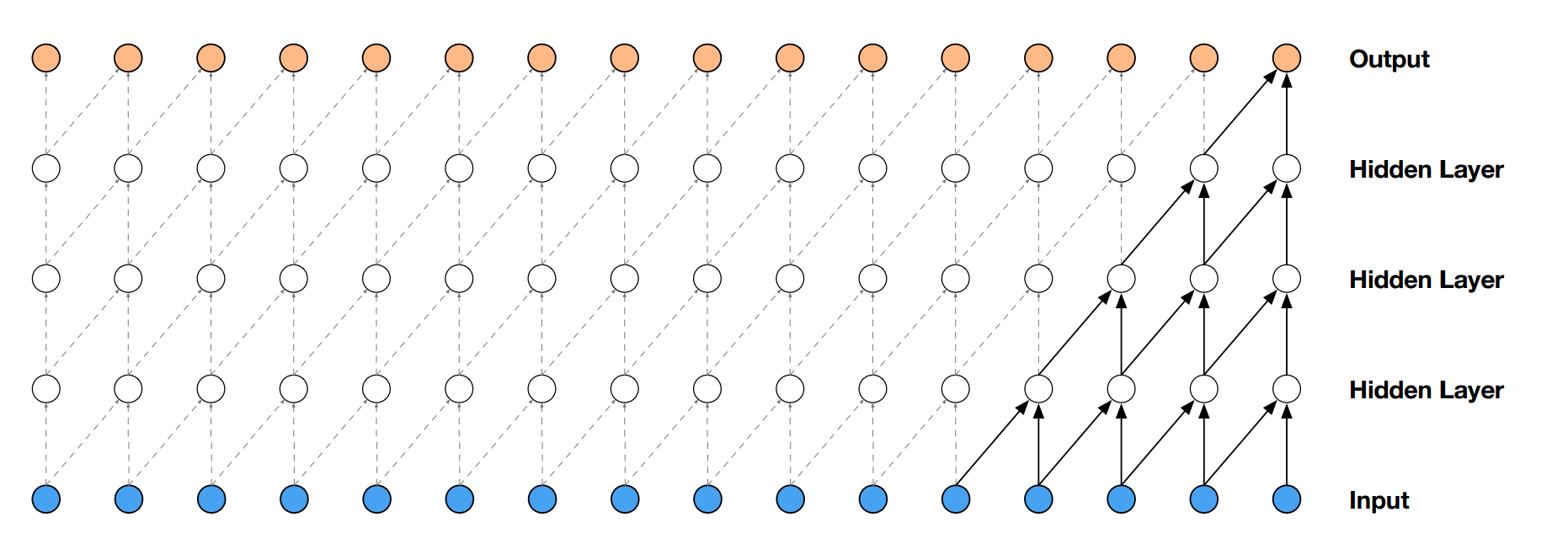
WaveNet: A Generative Model for Raw Audioより引用
それに対し,Dilated Convolution(atrous convolutionやconvolution weith holesともよばれます)は読み取る場所をずらしたところからうけとります.例えばDilation=4の場合,4だけ離れた位置から情報を受け取ります.このDilationを倍々にしていき,カーネルサイズを2とした場合,Dだけ離れた位置の情報を受取るには \(\log_2 D\)層だけ必要になります.今回のDが数百から数万の場合,10から20層程度あれば済むことになります.
今回はこのDilated Convolutionを使うことで遠距離にある情報を考慮できるモデルを作成します.

7.4.3. ブロック¶
それでは最初に,ネットワークの全体を設計します. このネットワークは二つのブロックから構成されます.
1つ目のブロックは長さが\(2^{17}\)の配列を入力として長さが\(2^{10}\)のベクトルを出力とします.これにより入力の128 (\(=2^{17}/2^{10}\))bpが出力の1つの位置に対応するようになります.これを実現しているのが,SqueezeBlockです.すなわち,SqueezeBlockは長さ131072bpからなるDNAの塩基配列を入力として受け取り,各フラグメントの長さに相当する128bp毎の情報が一つの値となるような畳込み処理を行います.結果として131072/128=1024の長さのベクトル列が出力されます.このベクトル列はフラグメント毎の特徴が一つのベクトルに圧縮されたものとみなすことができます.
二つ目のブロックは遠距離にある情報を考慮して各ベクトルの値を計算していく部分であり,DilatedBlockが担当します.DilatedBlockは,SqueezeBlockから出力された1024の長さのベクトル列を受け取り,Dilated Convolutionの仕組みを使うことで互いに離れた位置の情報を効率的に考慮した上で処理していき,入力と同じ1024の長さの出力を返します.この出力が,フラグメント毎に与えられたDNA関連タンパク質の結合可能性を表す数値(カバレッジ値)と一致するように学習を進めます.
それでは,以下のコードを実行してみましょう.
[6]:
import chainer
import chainer.functions as F
import chainer.links as L
import cupy as cp
bc = 24 # base channel
default_squeeze_params = [
# out_ch, kernel, stride, dropout
[bc*2, 21, 2, 0], #1 128 -> 64
[int(bc*2.5), 7, 4, 0.05], #2 64 -> 16
[int(bc*3.2), 7, 4, 0.05], #3 16 -> 4
[bc*4, 7, 4, 0.05] #4 4 -> 1
]
default_dilated_params = [
# out_ch, kernel, dilated, dropout
[bc, 3, 1, 0.1],
[bc, 3, 2, 0.1],
[bc, 3, 4, 0.1],
[bc, 3, 8, 0.1],
[bc, 3, 16, 0.1],
[bc, 3, 32, 0.1],
[bc, 3, 64, 0.1]
]
class Net(chainer.Chain):
def __init__(self, squeeze_params=default_squeeze_params, dilated_params=default_dilated_params, n_targets=10):
super(Net, self).__init__()
self._n_squeeze = len(squeeze_params)
self._n_dilated = len(dilated_params)
with self.init_scope():
in_ch = 4
for i, param in enumerate(squeeze_params):
out_ch, kernel, stride, do_rate = param
setattr(self, "s_{}".format(i), SqueezeBlock(in_ch, out_ch, kernel, stride, do_rate))
in_ch = out_ch
for i, param in enumerate(dilated_params):
out_ch, kernel, dilated, do_rate = param
setattr(self, "d_{}".format(i), DilatedBlock(in_ch, out_ch, kernel, dilated, do_rate))
in_ch += out_ch
self.l = L.ConvolutionND(1, None, n_targets, 1)
def forward(self, x):
# x : (B, X, 4)
xp = cp.get_array_module(x)
h = xp.transpose(x, (0, 2, 1))
h = h.astype(xp.float32)
for i in range(self._n_squeeze):
h = self["s_{}".format(i)](h)
hs = [h]
for i in range(self._n_dilated):
h = self["d_{}".format(i)](hs)
hs.append(h)
h = self.l(F.concat(hs, axis=1))
h = xp.transpose(h, (0, 2, 1))
return h
このネットワークは初期化時の引数としてSqueezeBlockに関するパラメータと,DilatedBlockに関するパラメータを受け取ります.
それぞれ,出力チャンネル,カーネルサイズ,プーリング,ドロップアウト率の四つ組からなるリストと,出力チャンネル,カーネルサイズ,dilatedサイズ・ドロップアウト率の四つ組からなるリストを受け取ります.
次に,ブロックの定義をします.
[7]:
import chainer
import chainer.functions as F
import chainer.links as L
import cupy as cp
class WNConvolutionND(L.ConvolutionND):
def __init__(self, *args, **kwargs):
super(WNConvolutionND, self).__init__(*args, **kwargs)
self.add_param('g', self.W.data.shape[0])
norm = np.linalg.norm(self.W.data.reshape(
self.W.data.shape[0], -1), axis=1)
self.g.data[...] = norm
def __call__(self, x):
norm = F.batch_l2_norm_squared(self.W) ** 0.5
channel_size = self.W.data.shape[0]
norm_broadcasted = F.broadcast_to(
F.reshape(norm, (channel_size, 1, 1)), self.W.data.shape)
g_broadcasted = F.broadcast_to(
F.reshape(self.g, (channel_size, 1, 1)), self.W.data.shape)
return F.convolution_nd(
x, g_broadcasted * self.W / norm_broadcasted, self.b, self.stride,
self.pad, self.cover_all, self.dilate)
class SqueezeBlock(chainer.Chain):
def __init__(self, in_ch, out_ch, kernel, stride, do_rate):
super(SqueezeBlock, self).__init__()
self.do_rate = do_rate
with self.init_scope():
pad = kernel // 2
self.conv = WNConvolutionND(1, in_ch, out_ch*2, kernel, pad=pad, stride=stride)
def forward(self, x):
h = self.conv(x)
h, g = F.split_axis(h, 2, 1)
h = F.dropout(h * F.sigmoid(g), self.do_rate)
return h
class DilatedBlock(chainer.Chain):
def __init__(self, in_ch, out_ch, kernel, dilate, do_rate):
super(DilatedBlock, self).__init__()
self.do_rate = do_rate
with self.init_scope():
self.conv = WNConvolutionND(1, in_ch, out_ch*2, kernel, pad=dilate, dilate=dilate)
def forward(self, xs):
x = F.concat(xs, axis=1)
h = self.conv(x)
h, g = F.split_axis(h, 2, 1)
h = F.dropout(h * F.sigmoid(g), self.do_rate)
return h

WeightNormalization[2]はパラメータの表現を長さと向きに分解して表現する手法で,今回の系列問題のような場合に使われる正規化法です.コード中ではWeightNormalizationが適用された畳み込み層であるWNConvolutionNDが定義されています.
SqueezeBlockは配列を縮めていき,長さが\(2^{17}\)の配列を\(2^{10}\)に縮めるためのブロックです(上図). 1次元配列を扱うためWNConvolutionNDを使い,最初の引数で1次元配列であることを示す1を指定しています. また,活性化関数では\(h = Wx * sigmoid(Ux)\)と表されるGated Linear
Unit[3]を利用しています.計算では効率化のため,WxとUxを別々に計算するのではなく2倍の出力チャンネル数を持つConvolutionを適用した後に出力結果をチャンネル方向に2つに分割し\((Wx, Ux)\),片方にsigmoid関数を適用した後,それらを要素毎にかけ合わせます.
DilatedBlockはすでに長さ1024の長さになった配列に対し,Dilated Convolutionを使って遠距離にある情報も使って計算していくブロックです(上図).引数としてdilatedを受け取ります.Dilated Convolutionを使う場合は通常のConvolution層(今回はConvolutionNDだが,Convolution2Dも同様)の引数にdilatedを加えるだけで計算できます.
また,DilatedBlockではDenseNet[4]と呼ばれる,以前の途中結果が全て次の層の入力として使われる手法を採用します(DilatedBlock内 forward()内のconcatがそれに対応).これはニューラルネットワークで多くのスキップ接続を作ることで,層が増えても勾配が減衰せず,学習がしやすくなることを利用したものです.
それでは,試しにネットワークを構築して,そこにサンプルデータを流してみましょう.
[8]:
import numpy as np
n = Net()
size = 131072 # 128 * 1024
batchsize = 4
x = np.empty((batchsize, size, 4), dtype=np.bool)
y = n.forward(x)
print(y.shape)
(4, 1024, 10)
(4, 1024, 10)
ここで,もともとバッチサイズ(B)=4, 入力長(L)=131072, 入力チャンネル数(C)=4だった配列が計算後はB=4, L=1024, C=10の配列となりました.
リンクテキスト今回予測するカバレッジ値は,フラグメント毎にDNA関連タンパク質がどの程度の頻度で結合したかを表すカウントデータであるとみなせます.そこで学習ではカウントデータに対する損失関数である対数ポアソン損失関数を利用します.
対数ポアソン損失関数を使う場合,モデルはポアソン分布の唯一のパラメータである平均を予測し,その予測された平均をもったポアソン分布を使った場合の学習データの尤度を計算します.そしてその尤度の最大化,それと同じである負の対数尤度の最小化を行います.この際,プログラム上では学習対象パラメータが含まれない項を無視しています. なお,この関数の最小値はそのままだと\(0\)にはならので,最小値である\(t \log t\)をあらかじめひいておき,損失関数の最小値が\(0\)となるようにします.
[9]:
import chainer.functions as F
import math
import sklearn
import numpy as np
def log_poisson_loss(log_x, t):
loss = F.mean(F.exp(log_x) - t * log_x)
t = chainer.cuda.to_cpu(t.astype(np.float32))
offset = F.mean(cp.array(t - t * np.ma.log(t)))
return loss - offset
def log_r2_score(log_x, t):
return F.r2_score(F.exp(log_x), t)
また,学習率の調整にCosineSchedulerを使います.ニューラルネットワークの学習では,徐々に学習率を小さくしていくと,より汎化性能の高い解を見つけられることがわかっています.ニューラルネットワークの学習の目的関数は多くの性能の悪い局所解があるため,最初は学習率を高くして局所解にはまらないようにして全体の中での良い解を探し,後半は徐々に学習率を0に近づけていき収束させるというものです. CosineSchedulerはCosine関数の0度から90度までの変化のように学習率を変化させます.また学習は初期が不安定なので最初のn_warmup回,学習率を0から初期学習率まで線形に増やすことも一般的です.今回は学習率が低めで学習も安定しているのでn_warmupは0としてあります.
[10]:
from chainer import training
import numpy as np
import math
class CosineScheduler(training.Extension):
def __init__(self, attr='lr', init_val=0.0001, n_decays=200, n_warmups=3, target=None, optimizer=None):
self._attr = attr
self._target = target
self._optimizer = optimizer
self._min_loss = None
self._last_value = None
self._init_val = init_val
self._n_decays = n_decays - n_warmups
self._decay_count = 0
self._n_warmups = n_warmups
def __call__(self, trainer):
updater = trainer.updater
optimizer = self._get_optimizer(trainer)
epoch = updater.epoch
if epoch < self._n_warmups:
value = self._init_val / (self._n_warmups + 1) * (epoch + 1)
else:
value = 0.5 * self._init_val * (1 + math.cos(math.pi * (epoch - self._n_warmups) / self._n_decays))
self._update_value(optimizer, value)
def _get_optimizer(self, trainer):
return self._optimizer or trainer.updater.get_optimizer('main')
def _update_value(self, optimizer, value):
setattr(optimizer, self._attr, value)
self._last_value = value
最後に学習中に訓練データに意味を変えない変化を加えるData Augmentationを適用します.これは画像において回転させたり,平行移動させたりする場合と同じです. 今回は128bp毎にカバレッジ値を予測していますが,数塩基(例えば4~8など)移動したとしてもカバレッジ値は同じ程度になることが期待されます.そこで最大max_shift分だけ配列を前後にシフトします(完全にランダムな塩基配列を余った部分に入れると実際の塩基配列の分布と変わる可能性があるのでここではroll()関数を巡回シフトしています).
[11]:
import chainer
import random
class PreprocessedDataset(chainer.dataset.DatasetMixin):
def __init__(self, xs, ys, max_shift):
self.xs = xs
self.ys = ys
self.max_shift = max_shift
def __len__(self):
return len(self.xs)
def get_example(self, i):
# It applies following preprocesses:
# - Cropping
# - Random flip
x = self.xs[i]
y = self.ys[i]
s = random.randint(-self.max_shift, self.max_shift)
x = np.roll(x, s, axis=0)
return x, y
これで全部準備ができました.残りはChainerのTrainerを改造して学習するだけです.以下のコードを実行してください.
元々のデータ全体では学習に時間がかかるので,データ/ratio分だけを学習,検証用データとして利用します.今回ratioは1に設定されています.この場合30分程度で学習が完了します.短い時間で試したい方はratio=1をratio=10やratio=20として実験してみてください.
[12]:
import chainer
import chainer.functions as F
import chainer.links as L
import numpy as np
from chainer.training import extensions
from chainer import training
import h5py
ml_h5 = h5py.File('seq.h5')
train_x = ml_h5['train_in']
train_y = ml_h5['train_out']
valid_x = ml_h5['valid_in']
valid_y = ml_h5['valid_out']
test_x = ml_h5['test_in']
test_y = ml_h5['test_out']
ratio = 1
train_x = train_x[:len(train_x)//ratio]
train_y = train_y[:len(train_y)//ratio]
valid_x = valid_x[:len(valid_x)//ratio]
valid_y = valid_y[:len(valid_y)//ratio]
max_shift_for_data_augmentation = 5
train = PreprocessedDataset(train_x, train_y, max_shift_for_data_augmentation)
val = chainer.datasets.TupleDataset(valid_x, valid_y)
batchsize = 8
train_iter = chainer.iterators.SerialIterator(train, batchsize)
val_iter = chainer.iterators.SerialIterator(val, batchsize, repeat=False, shuffle=False)
model = L.Classifier(Net(), lossfun=log_poisson_loss, accfun=log_r2_score)
lr = 0.001
optimizer = chainer.optimizers.Adam(alpha=lr, beta1=0.97, beta2=0.98)
optimizer.setup(model)
optimizer.add_hook(chainer.optimizer_hooks.GradientClipping(threshold=0.01))
updater = training.updaters.StandardUpdater(
train_iter, optimizer, device=0)
n_epochs = 10
n_warmups = 0
out = "out"
trainer = training.Trainer(updater, (n_epochs, 'epoch'), out=out)
trainer.extend(CosineScheduler(attr='alpha', init_val=lr, n_decays=n_epochs, n_warmups=n_warmups), trigger=(1, 'epoch'))
trainer.extend(extensions.Evaluator(val_iter, model, device = 0))
trainer.extend(extensions.LogReport(trigger=(0.2, 'epoch')))
trainer.extend(extensions.snapshot_object(model, 'model_epoch_{.updater.epoch}'), trigger=(1, 'epoch'))
trainer.extend(extensions.PrintReport(
['epoch', 'main/loss', 'validation/main/loss', 'elapsed_time']), trigger = (0.1, 'epoch'))
# trainer.extend(extensions.ProgressBar())
trainer.run()
epoch main/loss validation/main/loss elapsed_time
0 2.48903 67.7519
0 1.84639 117.127
0 1.89686 166.72
0 1.81704 215.449
1 1.85827 1.85512 274.106
1 1.81286 323.281
1 1.74802 372.488
1 1.80567 421.261
1 1.7467 470.755
2 1.70371 1.78047 528.83
2 1.77928 577.477
2 1.67051 626.814
2 1.6415 675.927
2 1.67238 725.017
3 1.69656 1.70897 782.987
3 1.63935 831.673
3 1.64996 881.092
3 1.63925 930.107
3 1.71683 979.111
4 1.63116 1.71748 1036.98
4 1.64786 1085.9
4 1.6442 1134.54
4 1.57821 1183.92
4 1.62886 1232.91
5 1.61523 1.66392 1290.8
5 1.65216 1339.78
5 1.61142 1388.37
5 1.61483 1437.71
5 1.57835 1486.61
6 1.56529 1.63406 1544.53
6 1.59062 1593.49
6 1.61102 1642.09
6 1.60003 1691.49
6 1.57222 1740.46
7 1.55098 1.62176 1798.31
7 1.54207 1847.28
7 1.5653 1895.92
7 1.57523 1944.68
7 1.61043 1993.73
8 1.57391 1.62377 2051.65
8 1.51835 2100.61
8 1.58225 2149.57
8 1.59289 2198.5
8 1.56643 2247.32
9 1.55151 1.62115 2305.72
9 1.53593 2354.7
9 1.57812 2403.76
9 1.54277 2452.85
9 1.55514 2501.51
学習が成功したならば,ディレクトリのout以下に学習されたモデルが出力されているはずです.実際にモデルが出力されているのかを確認しましょう.
[13]:
!ls -l out/
total 14172
-rw-r--r-- 1 root root 10080 Dec 16 05:24 log
-rw-r--r-- 1 root root 1445890 Dec 16 04:47 model_epoch_1
-rw-r--r-- 1 root root 1447626 Dec 16 05:25 model_epoch_10
-rw-r--r-- 1 root root 1446428 Dec 16 04:51 model_epoch_2
-rw-r--r-- 1 root root 1446742 Dec 16 04:55 model_epoch_3
-rw-r--r-- 1 root root 1447061 Dec 16 04:59 model_epoch_4
-rw-r--r-- 1 root root 1447268 Dec 16 05:04 model_epoch_5
-rw-r--r-- 1 root root 1447473 Dec 16 05:08 model_epoch_6
-rw-r--r-- 1 root root 1447585 Dec 16 05:12 model_epoch_7
-rw-r--r-- 1 root root 1447649 Dec 16 05:16 model_epoch_8
-rw-r--r-- 1 root root 1447650 Dec 16 05:21 model_epoch_9
次に,学習したモデルを用いてテストデータに対しても予測してみます.次のようにして学習が終わったモデルを読み込み,テストデータに対してモデルを適用してみましょう.
[14]:
import chainer
import chainer.links as L
%matplotlib inline
import matplotlib.pyplot as plt
model_n_epoch = 10
out_dir = 'out'
model = L.Classifier(Net())
chainer.serializers.load_npz('{}/model_epoch_{}'.format(out_dir, model_n_epoch), model)
predictor = model.predictor
print(len(test_x))
with chainer.no_backprop_mode():
test_y_estimated = F.exp(predictor(test_x[:1]))
test_y = test_y[:1]
print(test_y_estimated.shape)
print(test_y_estimated[0,:,0])
500
(1, 1024, 10)
variable([1.8674504 2.004048 1.68377 ... 0.81418294 0.7608197
0.8720923 ])
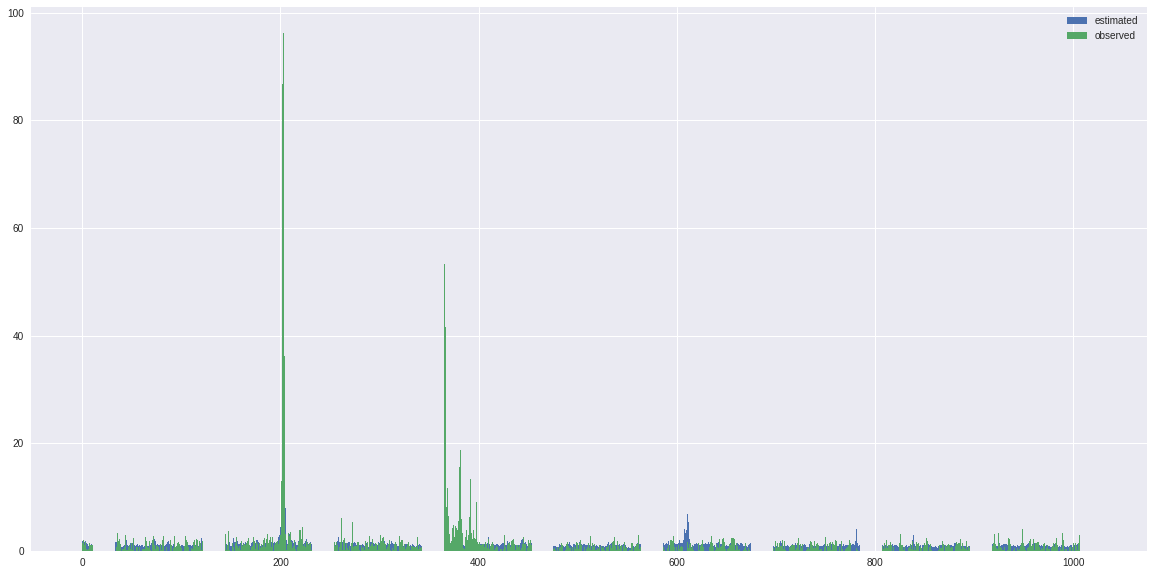
結果を抜粋して表示してみましょう.ここでは1つ目(i=0)の出力について正解と推定結果を出力しています.今回の場合でも,学習データを絞り(クラス数を10とした),学習回数も少ないですが,ピークを捉えられていることがわかると思います.
[15]:
y = test_y_estimated.data
fig_size = plt.rcParams["figure.figsize"]
fig_size[0] = 20
fig_size[1] = 10
i = 0
b1 = plt.bar(range(y.shape[1]), y[0,:,i])
b2 = plt.bar(range(y.shape[1]), test_y[0,:,i])
plt.legend((b1, b2), ('estimated', 'observed'))
[15]:
<matplotlib.legend.Legend at 0x7f037ea9f6d8>
時間に余裕があれば学習のn_epochsを10から30~50程度に増やしたり,層数を増やしたり,チャンネル数を増やしたりして,より高精度なモデルが学習できるのかを調べてみましょう.
- [1] "Sequential regulatory activity prediction across chromosomes with convolutional neural networks", D. R. Kelly and et al., Genome Res. 2018. 28: 739-750
- [2] "Weight Normalization: A Simple Reparameterization to Accelerate Training of Deep Neural Networks", T. Salimans and et al., arXiv:1602.07868
- [3] "Language Modeling with Gated Convolutional Networks", Y. N. Dauphin and et al., arXiv:1612.08083
- [4] "Densely Connected Convolutional Networks", G. Huang, and et al., CVPR 2017